COMPROMISO RENAL TEMPRANO EN NIÑA CON ENFERMEDAD DE FABRY CLÁSICA. REPORTE DE UN CASO.
Autores:
Fernando Perretta ¹,
Norberto Antongiovanni ²,
Sebastián Jaurretche ³
¹ Servicio de Terapia Intensiva del Hospital Dr. Enrique Erill de Escobar, Provincia de Buenos Aires, Argentina.
² Centro de Infusión y Estudio de Enfermedades Lisosomales del Instituto de Nefrología Clínica Pergamino, Provincia de Buenos Aires, Argentina.
³ Unidad de Enfermedades Lisosomales del Grupo Gamma Rosario, Provincia de Santa Fe, Argentina.
¹ˑ²ˑ³ Miembros de GINEF Argentina (Grupo de Investigación Nefrológica de la Enfermedad de Fabry).
Correspondencia:
Alsina 741, Pilar (CP: B1629), Provincia de Buenos Aires, Argentina
Teléfono: +54-91141789159
Fax: +54-2304423820
E-mail: fjperretta@hotmail.com
RESUMEN
La enfermedad de Fabry es un trastorno de depósito lisosomal ligado al cromosoma X, resultante de la deficiencia o ausencia de la enzima alfa galactosidasa A, que provoca la acumulación de glicoesfingolípidos complejos, principalmente globotriaosilceramida y sus metabolitos. Las manifestaciones de la EF son multisistémicas y comienzan en la infancia, alcanzando una afectación severa en la edad adulta. Se pueden observar las características acroparestesias en manos y pies, síntomas gastrointestinales, angioqueratomas, dishidrosis, pérdida de la audición, arritmias, miocardiopatía hipertrófica, accidentes cerebrovasculares e insuficiencia renal. Se presenta el caso de una niña con enfermedad de Fabry clásica y su compromiso renal incipiente.
Palabras clave: Enfermedad de Fabry; Alfa galactosidasa A; Globotriaosilceramida; Compromiso renal.
ABSTRACT
Fabry disease is an X-linked lysosomal storage disorder resulting from the deficiency or absence of the enzyme alpha galactosidase A, this defect leads to the systemic accumulation of globotriaosylceramide and its metabolites. The manifestations of FD are multisystemic and begin in early childhood, reaching a severe compromise in adulthood. Typical acroparesthesia in hands and feet, gastrointestinal symptoms, angiokeratomas, dyshidrosis, hearing loss, arrhythmias, hypertrophic cardiomyopathy, cerebrovascular accidents and renal failure can be observed. A case of incipient renal involvement in a girl with classic Fabry disease is reported.
KEYWORDS: Fabry disease; Alpha galactosidase A; Globotriaosylceramide; Renal involvement.
Introducción
La enfermedad de Fabry (EF) es provocada por la acumulación lisosomal de glicoesfingolípidos complejos, principalmente globotriaosilceramida (Gb3) y sus metabolitos 1. Dicho depósito dispara cascadas fisiopatogénicas en el endotelio vascular y células de diferentes tejidos (cardíacas, renales y nerviosas, entre otras) que conducen a la muerte celular, con progresión a fibrosis y deterioro orgánico irreversible 2,3. El almacenamiento de Gb3 se debe a la actividad deficiente o nula de la α-galactosidasa A (α-galA, EC 3.2.1.22). El gen GLA, que codifica la α-galA, está ubicado en el cromosoma X (Xq22.1), por lo cual prácticamente todos los hombres portadores de una mutación genética (hemicigota) desarrollan la enfermedad, mientras que las mujeres (heterocigotas) presentan una amplia variabilidad en la severidad de su fenotipo, debido principalmente a la inactivación aleatoria de uno de los cromosomas X en cada una de sus células (hipótesis de Lyon) 4. La intensidad o grado de los síntomas va a depender, en mayor parte, de la actividad residual de la enzima α-galA.
Las manifestaciones de la EF son multisistémicas y comienzan en la infancia, alcanzando una afectación severa en la tercera o cuarta década de la vida. Los principales signos y síntomas de esta enfermedad son las características acroparestesias en manos y pies, trastornos gastrointestinales, angioqueratomas, dishidrosis, intolerancia al ejercicio y al calor, pérdida de la audición, arritmias, miocardiopatía hipertrófica, accidentes cerebrovasculares e insuficiencia renal 5,6.
La EF es panétnica y, dada su baja incidencia, no se ha podido describir con exactitud su prevalencia, que varía entre 1:40.000 hombres a 1:117.000 nacidos vivos 7,8. La gran variabilidad fenotípica junto con la sintomatología inespecífica dificultan significativamente el diagnóstico, al cual se llega en edades adultas cuando el compromiso orgánico ya está instalado.
Gracias a estudios sistemáticos de detección de EF en centros de nefrología y diálisis, se ha podido avanzar en la determinación de la prevalencia en la población dialítica. La misma es de un 0.33% en hombres, demostrando que el screening es una estrategia útil de detección en pacientes con daño renal crónico 9.
Es de destacar que una vez realizado el diagnóstico se puede trabajar en el armado del árbol genealógico, lo que permitirá identificar a los familiares afectados, y así detectar a los pacientes en edades más tempranas 10.
En los últimos años, se ha avanzado en el entendimiento de la fisiopatología del daño tisular en la EF, principalmente en el compromiso orgánico temprano por acumulación de Gb3, donde se pudo observar que en pacientes pediátricos asintomáticos ya se detectan alteraciones tisulares 11. En este reporte se describe el compromiso renal incipiente en una paciente pediátrica con EF clásica.
Caso Clínico
Paciente femenina de 9 años de edad con diagnóstico de EF a los 5 años, por árbol genealógico. Mutación c.1244T>C (p.L415P) en estado de heterocigosis. Dosaje de alfa galactosidasa en gota de sangre en papel de filtro 1.5 umol/l/h (valor de referencia ≥4.0).
Se trata de un paciente eutrófica, escolarizada, con pautas neuromadurativas acorde a su edad cronológica, y que realiza actividad física habitual sin inconvenientes. Al examen físico: signos vitales conservados, peso 52 kg, talla 151 cm, facie tosca, broncoespasmos aislados por lo que recibe salbutamol inhalatorio según necesidad, acroparestesias leves e intermitentes con buena respuesta a la administración de carbamazepina 200 mg/día, y aislados angioqueratomas periumbilicales Figura 1.
Estudios Complementarios
Laboratorio: Hemoglobina 12.5 g/dl, leucocitos 6.150 mm³, glucemia 100 mg/dl, urea 14 mg/dl, creatinina 0.34 mg/dl (Tasa de filtrado glomerular estimada por fórmula de Schwartz de 183 ml/min), sodio 144 mEq/l, potasio 4.2 mEq/l, albúmina 4.37 g/dl, colesterol total 198 mg/dl y triglicéridos 103 mg/dl. Orina completa normal, albuminuria 2.7 ug/min (valor de referencia de 0 a 15) y proteinuria 30 mg/24 hs (valor de referencia < 150). Dosaje de Lyso-Gb3 en sangre 69.9 nmol/l (valor de referencia < 1.2).
Electrocardiograma dentro de parámetros fisiológicos para su edad. Eco Doppler cardíaco con reflujo leve fisiológico tricuspídeo y pulmonar, resto dentro de la normalidad. Resonancia magnética nuclear de cerebro normal. Examen oftalmológico con lámpara de hendidura que mostró córnea verticillata en ambos ojos. Ecografía abdominal y renal normal. Audiometría tonal dentro de la normalidad.
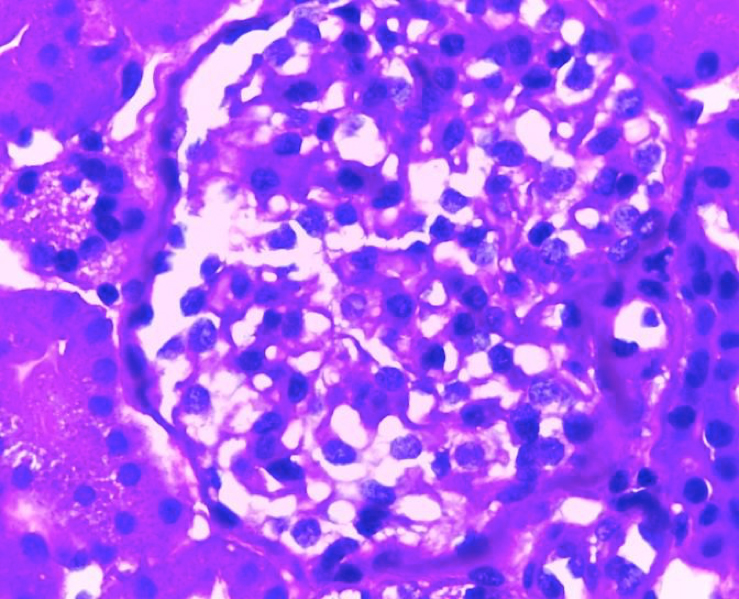
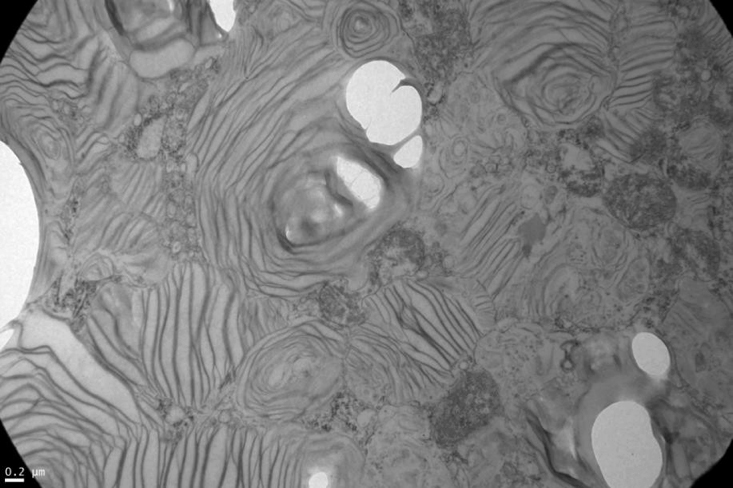
Si bien la paciente no presentaba datos clínicos de nefropatía (albuminuria/proteinuria), por la presencia de hiperfiltración glomerular asociada a su neuropatía periférica y a un dosaje elevado de Lyso-Gb3, se decide realizar punción biopsia renal (PBR). Microscopía óptica: luego de la coloración con hematoxlina-eosina, PAS, tricrómica de Masson y metenamina plata de Jones, se observan glomérulos con algún grado de clarificación del citoplasma podocitario que ocupa en promedio el 30% de los podocitos. Túbulos: algunos distales con clarificación y microvacuolización del citoplasma. Intersticio y vasos sin alteraciones Figura 2. Microscopía electrónica: en los cortes de un micrón se observaron glomérulos que presentaban microvacuolización podocitaria (Score 2 de la clasificación de Fogo). En túbulos leve clarificación en el citoplasma de proximales y distales. Intersticio y vasos sin alteraciones. Se observaron en el citoplasma de varios podocitos las características imágenes mieloides y en cebra que confirman el diagnóstico de la EF Figura 3. Luego de una evaluación multidisciplinaria de la paciente, se decidió iniciar terapia de reemplazo enzimático (TRE) con agalsidasa beta en dosis de 1 mg/kg de peso corporal cada 2 semanas por perfusión intravenosa.
DISCUSIÓN
Los depósitos de Gb3 se han encontrado en tejido placentario, lo que sugiere que el almacenamiento tisular ya está presente en el momento del nacimiento 12. Sin embargo, los niños típicamente no son sintomáticos en los primeros años de vida 11. Los pacientes habitualmente presentan en la infancia o la adolescencia síntomas que reflejan la pérdida progresiva de la función de pequeñas fibras nerviosas no mielinizadas del sistema somatosensorial y autónomo 11. Los primeros síntomas pueden incluir dolor neuropático crónico y/o ataques agudos de dolor (crisis Fabry); falta o disminución de la sudoración; tinnitus; intolerancia al frío, al calor o al ejercicio; trastornos gastrointestinales (por ejemplo, diarrea, malestar abdominal, náuseas y vómitos); y dificultad para ganar peso. Además, las lesiones cutáneas (angioqueratomas), las opacidades corneales sin afectación de la visión, y la presencia de proteinuria discreta (en varones adolescentes), están entre las primeras manifestaciones 13. Estos síntomas generalmente causan morbilidad a pesar de la ausencia de disfunción orgánica mayor, y además disminuyan significativamente la calidad de vida y el rendimiento de los niños en la escuela. Se ha reportado que el inicio de los síntomas puede ocurrir en la temprana infancia, antes de los 5 años 14.
El screening es una herramienta válida que puede ayudar a detectar pacientes con EF, y el armado del árbol genealógico como en este caso, a identificarlos en etapas más tempranas, antes que se produzca el daño orgánico 10.
La nefropatía es una de las principales complicaciones de la EF 10. Biopsias renales demuestran acumulación de Gb3 en células epiteliales tubulares, glomerulares y endoteliales, con glomérulo-esclerosis tan tempranamente como en la segunda década de vida e incluso en pacientes pediátricos sin albuminuria ni deterioro de la función renal 18,20. En la actualidad la única herramienta para detectar compromiso en etapa temprana es la PBR, pero su indicación de rutina es controversial, por los posibles riesgos del procedimiento, sobre todo en pacientes pediátricos. Es por ello que, el interés en los últimos años se ha centrado en el estudio de biomarcadores no invasivos capaces de detectar el daño renal temprano. El Lyso-Gb3 se ha relacionado con dolor neuropático y con cambios histológicos pre-albuminúricos, como en este caso. La medición plasmática de Lyso-Gb3 es valiosa para la confirmación del diagnóstico de la EF, particularmente en mujeres heterocigotas, y además sus valores se correlacionan con severidad de la enfermedad 15. Por otra parte, se está trabajando en la identificación de podocitos urinarios como potencial biomarcador de daño glomerular, tanto en las glomerulopatías primarias como secundarias 16. Específicamente en la EF se ha demostrado la presencia de podocituria y su asociación con la severidad de la nefropatía Fabry; observándose relación directa con la pérdida urinaria de proteínas e inversa con la tasa de filtrado glomerular 17. Aún, son necesarios nuevos biomarcadores que ayuden a la estratificación y cuantificación del daño renal en pacientes con EF, que puedan suplantar la PBR.
En el presente reporte describimos el compromiso histológico renal en una paciente pediátrica con EF clásica y signo-sintomatología inespecífica, previo a la pérdida urinaria de proteínas (albuminuria/proteinuria). En la PBR se describe un score 2 de la clasificación de Fogo 18; vacuolización podocitaria moderada. Este hallazgo confirma lo publicado por Camilla Tøndel en 2008, dónde afirma que los cambios glomerulares y vasculares están presentes antes de la proteinuria y de la caída del filtrado glomerular 19.También se ha descripto que el borramiento de los pies podocitarios en niños y niñas con EF clásica es un marcador temprano de nefropatía Fabry, antes de la presencia de albuminuria. Por tal motivo, las biopsias renales pueden ser esenciales en el diagnóstico precoz de la nefropatía, y también en la evaluación de la respuesta a la TRE 20. Es de destacar en esta paciente los valores de hiperfiltración renal que se pueden interpretar como un signo de compensación glomerular, a pesar de tener como limitante que los mismos fueron estimados por fórmula, y no medidos.
En este caso, la PBR aportó elementos útiles para el diagnóstico de la EF, confirmando la sospecha clínica. La carencia de biomarcadores no invasivos que se correlacionen con el grado de daño tisular determina que, a pesar de las controversias planteadas, la realización de PBR sea una intervención necesaria en determinados pacientes, incluso en edades pediátricas.
Desde 2001, la EF cuenta con un tratamiento específico, la TRE, la cual ha demostrado seguridad y eficacia en pacientes con EF 21. Ha sido descripto que las lesiones renales histológicas tempranas tienen respuesta favorable a la TRE cuando ésta se inicia de manera precoz. El clearence de Gb3 y la mejoría de las lesiones histológicas han sido confirmadas en PBR seriadas de pacientes pediátricos tratados con TRE, y tienen una correlación dosis-dependiente 19,20, observándose además re-acumulación luego del descenso de la dosis indicada 22.
Para concluir destacamos la importancia de sospechar la EF, su estudio exhaustivo, y el tratamiento precoz y oportuno, que cambiará la historia natural de esta enfermedad.
Conflictos de interés
Los autores declaran que no tienen conflictos de interés potenciales relacionados con los contenidos de este artículo.
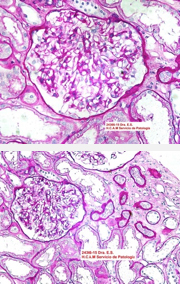
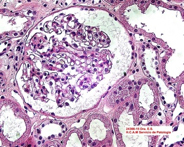
 Figura 1. Angioqueratomas periumbilicales.
Figura 1. Angioqueratomas periumbilicales.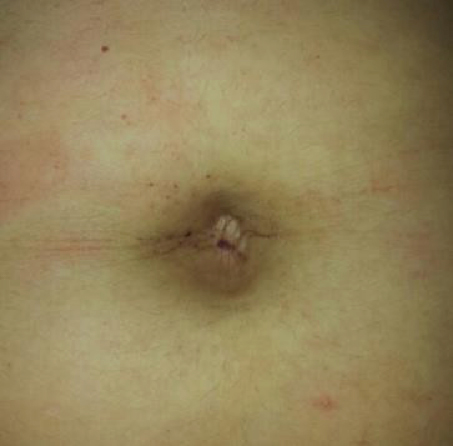
 Cuadro 1
Cuadro 1