Recomendaciones para el diagnóstico, tratamiento y seguimiento de la enfermedad de Fabry en Argentina.
Autores:
Politei J, Aiziczon D, Aguilar M, Alberton V, Alonso S, Amoreo O, Andrade L, Antongiovanni N, Arán MN, Arce P, Barán M, Bevione P, Biaiñ ME, Cabrera G, Chain J, Chiurchiu C, Cicerán A, Compte G, Cutrona R, De Arteaga J, Di Maio S, Di Pietrantonio S, Durand C, Fainboim A, Fernandez A, Figueredo H, Figueroa R, Frabasil J, Gaite J, Galarza M, Gomez Pizarro F, Guelbert N, Heguilen R, Jaurretche S, Kidd S, Lucca S, Luna M, Luna P, Marin I, Martinez A, Martinez L, Mazo G, Muraro J, Nieto E, Noli D, Ojeda P, Palacios C, Perez de Arenaza D, Pernasetti M, Perretta F, Raskovsky V, Recchia L, Risolo A, Rosa Diez G, Rugiero M, Schenone AB, Toniolo F, Trimarchi H, Vazquez Durand M, Vidoni J.
Resúmen
La enfermedad de Fabry es una esfingolipidosis lisosomal ligada al cromosoma X causada por mutaciones en el gen GLA con la resultante deficiencia de α-galactosidasa A. Las formas de presentación se dividen en un fenotipo "clásico" con inicio pediátrico y multisistémico, y un fenotipo "tardío" con manifestaciones principalmente cardíacas o renales. El patrón de inactivación del cromosoma X en las mujeres deriva en la presencia de sintomatología severa o formas atenuadas, lo que define la necesidad de tratamiento en algunas de ellas. La terapia de reemplazo enzimático, las chaperonas farmacológicas y los tratamientos de sostén resultarán en un beneficio clínico, dependiendo del momento en que sean indicados. A la fecha, gran parte de la literatura reporta los resultados luego de un inicio tardío del tratamiento, una vez que se ha producido un daño orgánico irreversible. Estas recomendaciones de un panel de expertos y tratantes de la enfermedad buscan desarrollar directrices actualizadas y específicas para un diagnóstico y tratamiento temprano, así como también para el seguimiento de los pacientes con enfermedad de Fabry en nuestro país.
Palabras clave: nefropatía por IgA, enfermedad de cambios mínimos, síndrome nefrótico.
ABSTRACTS
Fabry disease is a lysosomal X linked sphingolipidosis as a result of mutations in the GLA gene that leads to a deficiency of α-galactosidase A. The spectrum of presentation are divided into a "clas- sic" phenotype with pediatric and multisystemic manifestations, and a late onset phenotype with mainly cardiac or renal involvement. The pattern of inactivation of X chromosome in women de- rives in the presence of severe symptoms or attenuated forms, which defines the need for treat- ment in some of them. Enzyme replacement therapy, pharmacological chaperones and supportive treatments will result in a clinical benefit, depending on when they are indicated. To date, much of the literature reports results after a late treatment indication, once irreversible organic damage has occurred. These recommendations comming from a panel of disease experts and treaters seek to develop updated and specific guidelines for the diagnosis, treatment and follow-up of patients with Fabry disease in our country.
Key words:Fabry disease, Genetics, Lysosomal, Enzyme Replacement Therapy
Introducción
La enfermedad de Fabry (OMIM 301500) es el resultado de la deficiencia total o parcial de la enzima lisosomal α-galactosidasa A (α-galA), que resulta en el acúmulo patológico de glicoesfingolípidos (principalmente globotriaosilceramida -Gl3- y su forma deacilada globotriaosilesfingosina -LisoGl3-) en paredes vasculares, células diferenciadas (cardiomiocitos, podocitos, etc) y fluidos (sangre, orina) 1. La deficiencia enzimática es la expresión de mutaciones en el gen GLA, que se encuentra localizado en el brazo largo del cromosoma X 2.
Aun cuando originalmente fue descripta como una enfermedad cuyos síntomas de inicio se presentan en la primera década de vida (dolor neuropático en manos y pies, compromiso gastrointestinal, angioqueratomas, etc), en la actualidad sabemos que esta forma de presentación corresponde a la forma Clásica (Tipo 1), y que existe una forma tardía o del adulto (Tipo 2) donde estos síntomas están ausentes 1. Recientes análisis de algunos resultados internacionales de pesquisa neonatal muestran una frecuencia de 1 en 22,570 en hombres con formas clásicas y 1 en 1390 hombres en formas tardías, lo que posicionaría a esta patología como la enfermedad de depósito lisosomal más frecuente 3.
La caracterización de los distintos fenotipos, la posibilidad de medir nuevos sustratos y el reconocimiento de daño tisular irreversible en la infancia ha llevado a cambiar el seguimiento y modificar las intervenciones terapéuticas en esta enfermedad 4. A la fecha disponemos de 16 años de experiencia con el tratamiento de reemplazo enzimático en Argentina, siendo que nuevos fármacos se han aprobado recientemente y varios se encuentran en etapa de investigación.
Estas recomendaciones son el resultado de una primera reunión presencial de un grupo de expertos y tratantes de la enfermedad de Fabry realizada el día 26 de mayo de 2018. Este grupo de colegas representa todas las especialidades relacionadas al manejo de la enfermedad: bioquímica, clínica médica, cardiología, dermatología, genética, nefrología, neurología, oftalmología, otorrinolaringología, pediatría y nefrología infantil. Luego de la primer reunión se distribuyó un manuscrito fuente que fue enviado y revisado por los participantes, quienes luego de realizar las modificaciones pertinentes arribaron a un consenso para su publicación. Estas recomendaciones son la expresión del conocimiento y experiencia de cada participante.
Signos y Síntomas
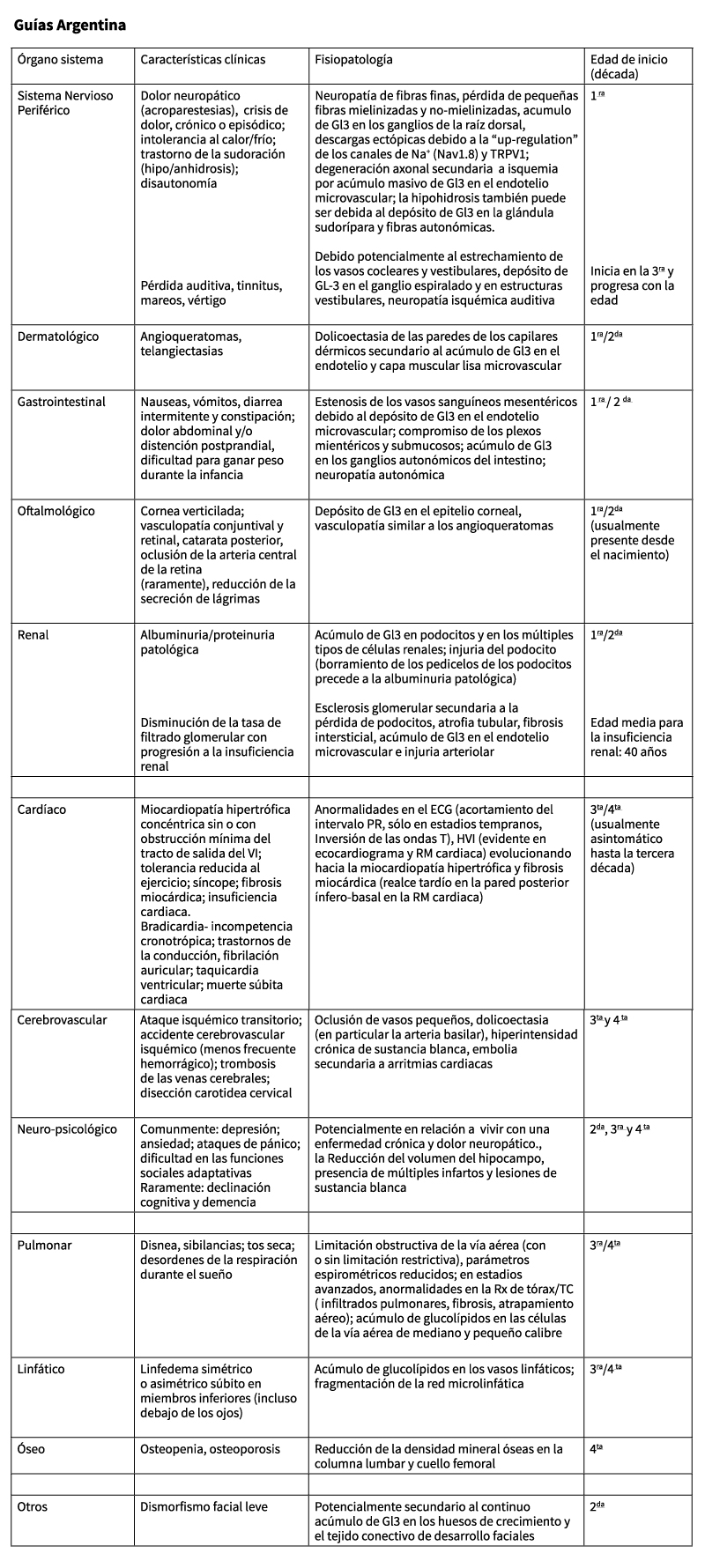
En la forma clásica (tipo 1) de la enfermedad las primeras manifestaciones se describen antes de los 7 años de edad como crisis de dolor neuropático en manos y pies (típicamente desencadenadas por el ejercicio, fiebre y cambios de temperatura), dolor abdominal post ingesta y diarrea frecuente 5. Así también, lesiones dermatológicas (angioqueratomas) e hipohidrosis pueden estar presentes antes de la segunda década de vida. Posteriormente la hipoacusia, acúfenos, vértigo y el compromiso ocular suelen acompañar al hallazgo de podocituria y proteinuria. Luego de la tercera década manifestaciones cardiacas como arritmias, dolor precordial, hipertrofia ventricular izquierda y accidentes cerebrovasculares pueden presentarse. La enfermedad renal crónica progresiva puede requerir terapias de reemplazo de la función renal luego de los 30 años de edad en esta variante clásica 1,2. Otras manifestaciones como compromiso respiratorio y osteoporosis han sido reportadas previamente 1.
Las formas de inicio del adulto (tipo 2) se caracterizan por el compromiso principalmente cardíaco (variante cardiaca) o renal (variante renal), con ausencia de las manifestaciones tempranas descriptas en los niños 6. Si bien existen reportes donde la córnea verticilada o la hipoacusia pueden estar presentes también en estas variantes tardías, no se comparan en severidad a lo descripto en la forma clásica 7.
Las mujeres con enfermedad de Fabry pueden manifestar la enfermedad desde formas muy poco sintomáticas hasta formas severas, similares a la descripta en los hombres 1,8. El concepto de "mujeres portadoras" en las enfermedades ligadas al cromosoma X ha sido reconsiderado, ya que la inactivación aleatoria de uno de los cromosomas X (fenómeno de Lionización) puede explicar la presencia o ausencia de sintomatología, en relación al porcentaje de células que expresan el cromosoma X mutado o sano 9. Datos recientes obtenidos del registro Fabry internacional confirman que los accidentes cerebrovasculares y el compromiso cardíaco son frecuentes en mujeres a una edad mayor a la reportada en hombres y el daño renal puede ser evidente a la misma edad media que los hombres, principalmente en las mujeres que expresan el cromosoma X mutado en la mayoría de sus células 8.
Aspectos Genéticos
A la fecha, más de 900 mutaciones en el gen GLA han sido reportadas en la literatura (10). El análisis de la secuencia del gen GLA por un especialista permite identificar la presencia de diferentes tipos de mutaciones: duplicaciones, inserciones, deleciones, cambios en el marco de lectura (frameshift) y mutaciones sin sentido (nonsense), entre otras 11. Todas las anteriores producirán una enzima truncada, incompleta y no funcional, lo que resultará en una actividad enzimática muy disminuida o nula y un fenotipo clásico. Por otro lado, algunas mutaciones con cambio de sentido (missense) pueden derivar en la producción de una enzima con actividad funcional residual, lo que explica la presencia de los fenotipos tardíos. Al mismo tiempo se han descripto mutaciones con cambio de sentido que resultan en actividad enzimática nula y se asocian a fenotipo clásico 12.
La inactivación aleatoria de uno de los cromosomas X en las mujeres (heterocigotas) es un fenómeno que puede explicar en parte la variabilidad fenotípica reportada. Actualmente es posible analizar el patrón de inactivación del cromosoma X, lo que brinda información y pronóstico para cada paciente heterocigota 13. En resumen, las mujeres que expresen en la mayoría de sus células el cromosoma X funcional (sin mutación) la presencia de síntomas sera escasa o aún ausente; por el contrario en mujeres que expresen el cromosoma X mutado sus síntomas y pronóstico serán similares a los descriptos en los hombres que porten esa mutación en la familia. Una de las limitaciones de este estudio es que el patrón de inactivación del cromosoma X puede diferir en los distintos tejidos de una misma paciente, dificultando su interpretación 9,13.
Las manifestaciones de la enfermedad de Fabry pueden ser diferentes en severidad aún en pacientes con la misma mutación, por lo que la correlación genotipo-fenotipo actualmente sigue siendo un desafío. Solo algunas pocas mutaciones como la p.N215S o la IVS4+919G>A muestran un correlato definido con manifestaciones primariamente cardiacas. Estas variantes cardíacas de inicio tardío ha ido en aumento desde que se ha incluido el gen GLA en el panel de genes para estudio de pacientes con miocardiopatía hipertrófica mediante el método de secuenciación masiva en paralelo 14-16. Existen mutaciones en el gen GLA que se consideran a priori variantes de significado incierto por tratarse de variantes de tipo novel o para las cuales no hay suficiente evidencia científica para clasificarlas como patogénicas o benignas 17. La posibilidad de cuantificar el LisoGl3 en plasma o la obtención y análisis de tejido por medio de biopsias suelen ser los métodos para comprobar la patogenicidad de estas mutaciones. Finalmente, se han definido en la actualidad un número importante de polimorfismos (variantes no patogénicas o benignas), donde la ausencia de biomarcadores en plasma y/o tejidos descartan la enfermedad 17. Estas variantes benignas deben ser descartadas antes de la prescripción del tratamiento.
METodoLoGíA diAGNÓSTiCA
La actividad enzimática de α-galA puede medirse en gotas de sangre en papel de filtro, leucocitos, plasma, y fibroblastos, siendo los dos primeros los tipos de muestra más utilizados 17,18.
Si bien el dosaje enzimático en leucocitos es considerado el método Gold Standard para el diagnóstico en varones, la presencia de variantes benignas que reducen la actividad enzimática in vitro, puede generar una interpretación errónea (resultado falso positivo). Otra limitación de la cuantificación de la actividad enzimática es el hallazgo de un resultado normal hasta en el 40% de las mujeres, debido a la inactivación aleatoria del cromosoma X 19. Por lo antedicho la identificación de una mutación patogénica mediante el estudio del gen GLA es relevante para realizar el diagnóstico en mujeres y confirmar el diagnóstico en hombres, descartando la presencia de variantes benignas. Por otro lado, conocer la mutación aporta en muchos casos información relacionada al fenotipo y permite realizar la búsqueda de la variante genética en otros familiares en riesgo.
Cuando una mutación no se encuentre descripta previamente en bases de datos o sea considerada de significado incierto (VUS), la presencia de sintomatología clásica (dolor neuropático, cornea verticilada, angioqueratomas, etc) junto con actividad enzimática descendida y niveles elevados del biomarcador LisoGl3, son herramientas suficientes para confirmar el diagnóstico de enfermedad de Fabry 17. Actualmente estudios en poblaciones de riesgo (servicios de hemodiálisis, pacientes con miocardiopatías hipertróficas, accidente cerebrovascular, etc) llevan a la descripción de variantes de significado incierto, donde la ausencia de síntomas característicos generan la duda entre variantes benignas o variantes asociadas a las formas del adulto (tardío) en el que el compromiso orgánico se limitará a un solo órgano 20. La posibilidad de realizar un dosaje de LisoGl3 en plasma u orina es una herramienta que puede confirmar el diagnóstico de enfermedad de Fabry. La presencia de valores elevados de LisoGl3 en plasma se observa en todos los varones con fenotipo clásico y fenotipo tardío, siendo que valores normales en hombres descartan la enfermedad. En mujeres con fenotipo clásico los niveles de LisoGl3 siempre se reportan elevados (aunque en menor nivel que en los hombres); la única limitación de este método es la posibilidad de encontrar valores normales en algunas mujeres con fenotipo tardío 21,22.
En casos puntuales, como ante la presencia de VUS o cuando los métodos actuales de secuenciación no permitan hallar la variante genética que ocasiona la deficiencia enzimática, la toma de biopsia cardiaca o renal con búsqueda de los respectivos depósitos intralisosomales de sustratos, puede ser la forma definitiva de confirmar la enfermedad 17.
TRATAMIENTO
El tratamiento de la enfermedad de Fabry puede ser dividido en una terapéutica específica (terapia de reemplazo enzimático o chaperonas farmacológicas) y un tratamiento de soporte sintomático. El tratamiento específico buscará prevenir la aparición de daño orgánico irreversible, al mismo tiempo que indicado en etapas tempranas podra revertir algunos mecanismos fisiopatológicos que finalizan en la muerte celular. El tratamiento debe estar supervisado por un equipo multidisciplinario de expertos en la enfermedad que incluya entre otros a pediatras (en caso de iniciar tratamiento en niños), clínicos, neurólogos, cardiólogos, nefrólogos, oftalmólogos, dermatólogos, otorrinolaringólogos y genetistas clínico. No existe ninguna especialidad por sobre la otra en cuanto a la decisión del tratamiento o monitoreo de la enfermedad.
TERAPiA dE REEMPLAzo ENziMáTiCo
Actualmente, en Argentina se encuentran disponibles dos formulaciones de terapia de reemplazo enzimático para la enfermedad de Fabry: agalsidasa alfa (Replagal®, Shire HGT, Inc., Cambridge, MA, USA) y agalsidasa beta (Fabrazyme®, Sanofi Genzyme, Cambridge, MA, USA). La agalsidasa alfa fue aprobada en varios países con una dosis de 0.2 mg/kg cada 14 días 23. A la fecha no ha recibido aprobación por la Food and Drug Administration (FDA) en Estados Unidos. La agalsidasa beta ha sido aprobada en todos los países (incluyendo Estados Unidos) con una dosis de 1mg/kg cada 14 días 24. Ambas formulaciones corresponden alfa galactosidasa recombinante humana.
iNiCio TEMPRANo dE LA TERAPiA dE REEMPLAzo ENziMáTiCo.
Existe evidencia científica que demuestra que un inicio temprano de la terapia de reemplazo enzimático se asocia a mejores resultados, tanto desde la respuesta clínica y bioquímica, como también en relación a la remoción de sustrato a nivel tisular 25. Estudios en pacientes pediátricos y jóvenes (< 33 años) a los que se realizó una biopsia renal al momento de iniciar el tratamiento y un control por biopsia luego de 5 y 10 años con distintas dosis de terapia de reemplazo enzimático confirmaron estos resultados 26. Si bien ambas formulaciones de terapia de reemplazo enzimático (agalsidasa alfa y beta) mostraron una rápida remoción de sustrato a nivel endotelial glomerular, sólo la dosis de 1 mg/kg de agalsidasa beta mostró una significativa remoción de Gl3 en podocitos, células diferenciadas cuya pérdida urinaria se relaciona en forma directa con la progresión a proteinuria e insuficiencia renal 26. Este respuesta dosis dependiente también fue evidente en cuanto a la disminución del índice proteinuria/creatininuria. Estos estudios han sido replicados por otros autores lo que confirma que la remoción de sustrato responde en forma directa con la dosis de agalsidasa 27-29
Los resultados positivos en relación a la masa ventricular fueron reportados en mayor cuantía cuando el tratamiento se inicia antes de los 40 años 30. Posiblemente, esta respuesta positiva en los pacientes más jóvenes esté relacionada al menor o ausente desarrollo de fibrosis miocárdica 31. Un reciente estudio de extensión a 10 años de tratamiento con agalsidasa beta a 1 mg/kg cada 14 días en los pacientes que participaron de la fase III (que llevó a su aprobación por parte de la FDA), mostró que iniciar el tratamiento en forma temprana con una función renal normal se asocia a mejores resultados cardiacos, renales y cerebrovasculares 32. Finalmente, un estudio que evaluó la incidencia de aparición de "eventos serios" renales, cardiacos y cerebrovasculares luego de 5 años de agalsidasa beta, concluyó que tanto en el grupo de pacientes de alto riesgo (hombres, > 40 años con daño orgánico pretratamiento) o bajo riesgo (hombres, < 40 años, sin eventos pretratamiento) la incidencia de eventos disminuyó significativamente luego de 6 meses de iniciado el tratamiento 33.
Si bien el LisoGl3 es una herramienta diagnóstica, sus niveles séricos son considerados un parámetro de severidad y su disminución durante el tratamiento refleja la eficacia del mismo. El efecto de la terapia de reemplazo enzimático en relación a la disminución del LisoGl3 fue estudiado y comparado en un grupo de pacientes que iniciaron el tratamiento antes de los 25 años o después. Los pacientes que iniciaron tratamiento antes de los 25 años mostraron una mayor caída del LisoGl3 en plasma en relación a quienes lo iniciaron después de los 25 años 25. Esto refleja que una mejor respuesta bioquímica se asocia al momento de inicio del tratamiento. Finalmente, la disminución del LisoGl3 plasmático ha mostrado una respuesta dosis dependiente, donde la dosis de 0,2 mg/kg de agalsidasa alfa se asocia a una reducción significativamente menor que con el uso de agalsidasa beta a 1mg/kg 34,35.
CRiTERioS PARA EL iNiCio dE LA TERAPiA dE REEMPLAzo ENziMáTiCo
El tratamiento con terapia de reemplazo enzimático requiere un diagnóstico confirmado de enfermedad de Fabry. Para una mejor comprensión los criterios de inicio de tratamiento se describen en relación al fenotipo, edad y a la presencia o no de síntomas característicos de cada paciente.
Tratamiento en pediatría.
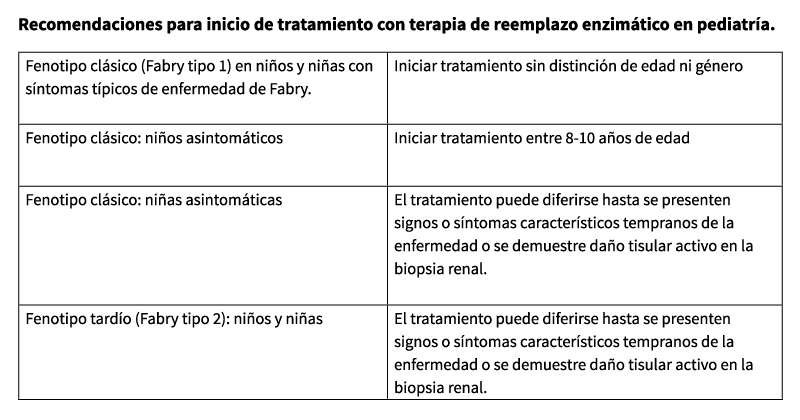
En pacientes pediátricos con fenotipo clásico y presencia de signos y síntomas característicos de la enfermedad de Fabry se recomienda iniciar terapia de reemplazo enzimático sin distinción de edad ni género 36.
En pacientes pediátricos varones con fenotipo clásico asintomáticos se recomienda iniciar tratamiento entre los 8-10 años de edad en base a los resultados de biopsias renales reportados en la literatura científica, donde todos los pacientes estudiados mostraron daño podocitario temprano 37,38. Reportes en la literatura indican la presencia de inclusiones podocitarias en biopsia renal de pacientes pediátricos de ambos sexos con fenotipo clásico que presentaban algún grado de acroparestesias como única manifestación de la enfermedad 28. Es por esto, que se recomienda no subestimar la presencia de síntomas característicos al momento de la decisión terapéutica, y si éstos no se evidenciaran de forma objetiva, la detección de signos histológicos mediante biopsia representa una herramienta de gran valor para confirmar el daño tisular e indicar el tratamiento. En pacientes pediátricas (niñas) con fenotipo clásico asintomáticas o varones/niñas con variantes del adulto la terapia de reemplazo enzimático puede diferirse hasta se presenten signos o síntomas característicos tempranos de la enfermedad o se demuestre daño tisular activo en la biopsia renal y/o cardiaca 36.
Tratamiento en adultos.
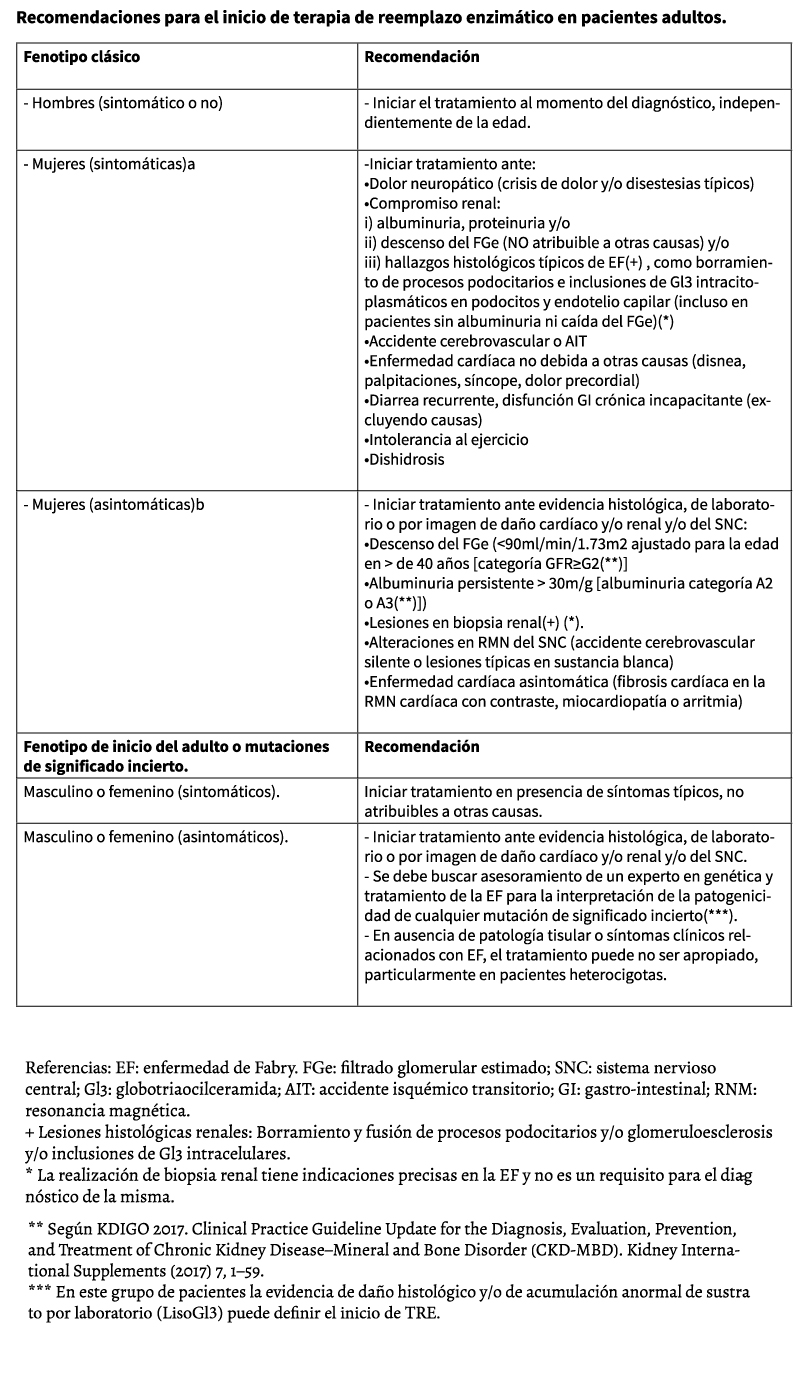
En pacientes hombres con fenotipo clásico, el tratamiento debe ser iniciado en edad pediátrica. Si el diagnóstico se realiza en edad adulta el tratamiento debe comenzar en forma inmediata 39.
En mujeres con fenotipo clásico, si existen síntomas característicos se debe iniciar tratamiento a la brevedad. Cuando no se evidencian síntomas en mujeres con fenotipo clásico, la presencia de signos de compromiso renal, cardiaco o cerebrovascular en el laboratorio, imágenes o biopsia, justifican iniciar el tratamiento 39.
En pacientes adultos con fenotipo de inicio del adulto (Fabry tipo 2), la terapia de reemplazo puede diferirse hasta que se presenten signos o síntomas característicos de la enfermedad o se demuestre daño tisular activo en la biopsia renal y/o cardiaca 40.
CRiTERioS PARA No iNiCiAR/SUSPENdER EL TRATAMiENTo
Estos criterios deben ser aplicados en pacientes hombres o mujeres, sin diferencias entre los fenotipos 40.
ChAPERoNAS FARMACoLÓGiCAS
La chaperona farmacológica migalastat (GalafoldTM; Amicus Therapeutics, Cranbury, NJ, USA) ha sido recientemente aprobada en Europa, Canada y Japón para su uso en pacientes portadores de mutaciones "respondedoras", en base a la respuesta de realce enzimático en células humanas renales de embrión con transfección de mutación en el gen GLA. La aprobación actual es solo para pacientes mayores a 16 años y que presenten un filtrado glomerular ≥ 30 ml/min 41.
Si bien el protocolo inicial a 6 meses de tratamiento no mostró resultado positivo respecto al punto de eficacia terminal principal contra placebo (disminución del 50% en el número de inclusiones de Gl3 en células endoteliales capilares del intersticio renal), se evidenció mejoría de la diarrea y reflujo, así como también una reducción de la masa ventricular izquierda 41,42.
Los autores de estas recomendaciones declaran no tener a la fecha experiencia con el uso de este tratamiento, aún no registrado ni aprobado en el país.
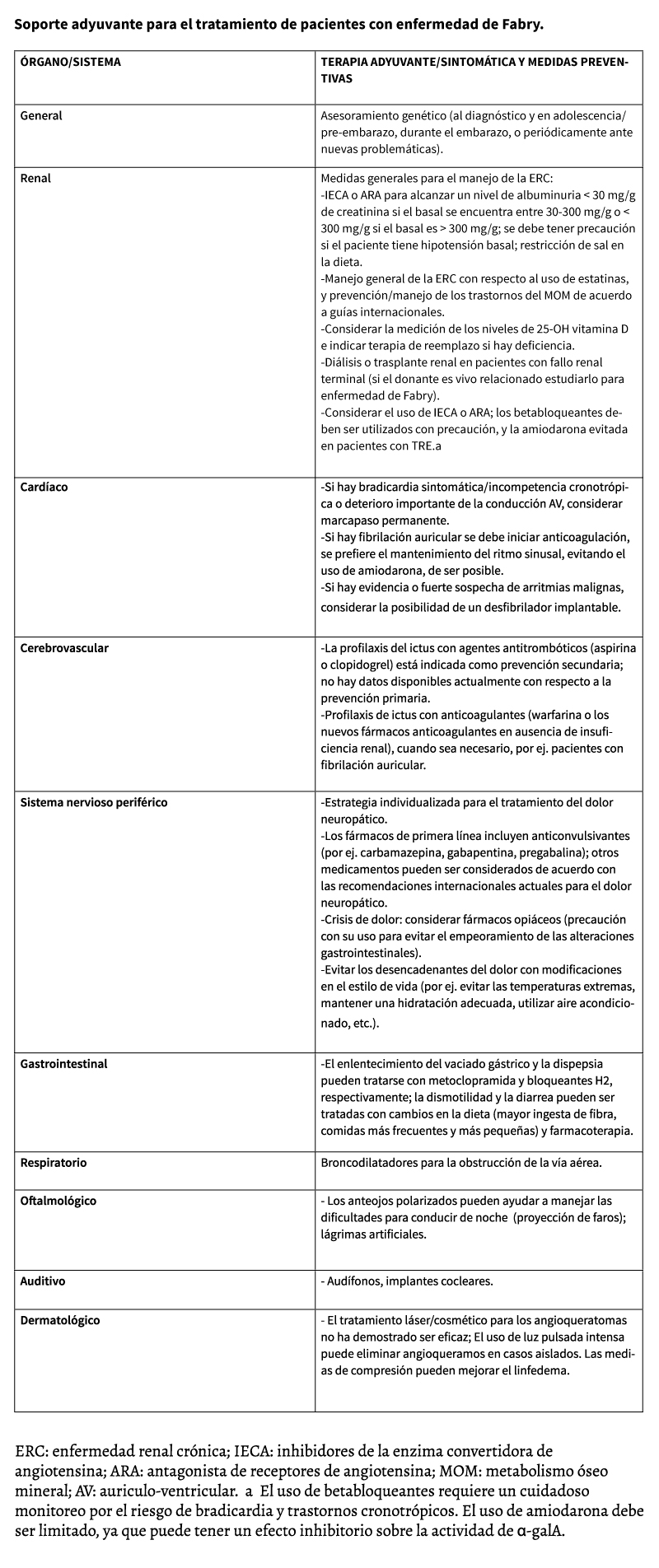
TRATAMiENTo dE SoPoRTE SiNToMáTiCo
El tratamiento específico para la enfermedad de Fabry requiere de un control estricto de los factores de riesgo cardiovascular, como también medidas farmacológicas para el manejo del dolor neuropático, compromiso gastrointestinal, control de la proteinuria, etc. Medidas preventivas como antiagregantes plaquetarios y modificaciones del estilo de vida entre otras, deben ser consideradas desde etapas precoces de la enfermedad 39.
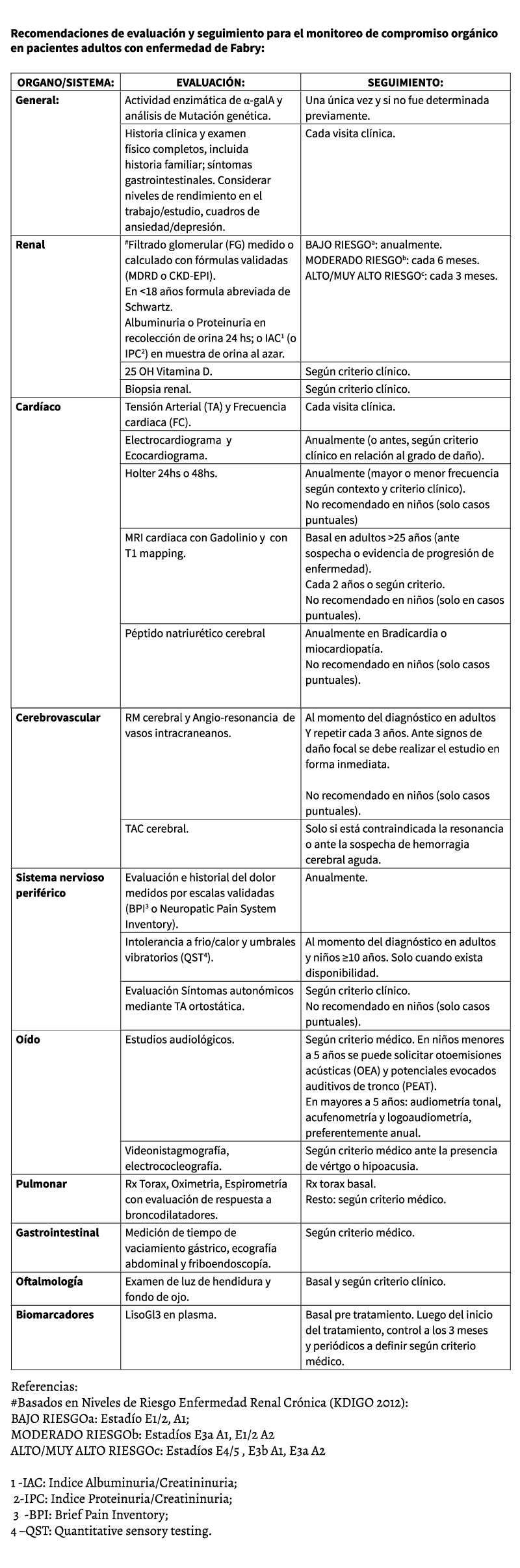
MoNiToREo dE LA ENFERMEdAd
El monitoreo de la enfermedad de Fabry debe ajustarse a distintas variables como la edad del paciente, el fenotipo y el grado de daño al momento del diagnóstico. Como guía recomendada se describen en la Tabla 6 el tipo de estudio a realizar y su necesidad de repetición en relación al órgano a monitorear.
PoBLACioNES ESPECiALES
Poblaciones en riesgo: se consideran como poblaciones de riesgo para realizar estudios de pesquisa (screening) a los pacientes en diálisis o en plan de trasplante renal (aún ya trasplantados), miocardiopatías hipertróficas y accidentes cerebrovasculares en jóvenes. Es fundamental conocer las manifestaciones tempranas de la enfermedad y los antecedentes familiares para realizar cuestionarios dirigidos que pueden llevar a elevar el índice de sospecha 20. Aun en ausencia de sintomatología clásica, las variantes del adulto deben ser sospechadas por nefrólogos y cardiólogos, ya que estas variantes pueden limitarse a un solo órgano.
Embarazo y Lactancia:
Los fármacos se encuentran clasificados en distintas categorías de riesgo según la FDA. La agalsidasa alfa y beta se pertenecen a la categoría B, donde los estudios de reproducción en animales no han determinado riesgo para el feto y no existen estudios controlados en embarazadas. Se dispone de reportes en la literatura con el uso de ambas agalsidasas donde no se constatan efectos adversos ni efectos deletéreos sobre el feto 43-46, por lo que se acepta su uso durante el embarazo. No se conoce si la agalsidasa es excretada en leche materna, por lo que no se han realizado recomendaciones definitivas respecto a la lactancia durante el tratamiento.
Pacientes en diálisis:
Se recomienda realizar las infusiones durante las sesiones de diálisis en pacientes con necesidad de terapia de reemplazo renal, debido a que el peso molecular de la agalsidasa no permite ser filtrada.
MANEjo PRáCTiCo dE LAS iNFUSioNES y dE LAS PoSiBLES REACCioNES ASoCiAdAS
Medicación antes de tratamiento, 1 hora preinfusión:
• Paracetamol 2 tabletas de 500 mg vía oral.
• Hidroxicina 1 tableta de 25 mg vía oral u otro antihistamínico similar según disponibilidad local.
• Control de signos vitales (presión arterial, frecuencia cardíaca y respiratoria, temperatura)
• En caso de que el paciente haya presentado fiebre o escalofríos durante infusiones previas, administrar ibuprofeno 1 tableta de 400 mg vía oral en las siguientes infusiones.
MANEjo dE REACCioNES dE hiPERSENSiBiLidAd
A. Reacciones leves, definidas como la presencia de:
• Sensación de calor.
• Urticaria menor o localizada (< 5% de superficie corporal).
• Hormigueos periféricos.
• Prurito cutáneo.
Conducta recomendada:
- Disminuir la velocidad de infusión a la mitad (registrar hora y velocidad de goteo).
- Administrar difenhidramina 50 mg, vía oral o intravenoso en 5-10 minutos.
- Si los síntomas desaparecen, volver a la velocidad de infusión previa en forma escalonada (en 15-30 minutos).
- Si la severidad de los síntomas aumenta, detener infusión.
B. Reacciones moderadas, definidas como la presencia de:
• Disnea leve.
• Urticaria generalizada.
• Náuseas y vómitos.
• Taquicardia.
• Prurito generalizado, calor y ansiedad.
• Enrojecimiento (flushing).
• Angioedema.
Conducta recomendada:
- Detener la infusión para evaluar la severidad o alternativamente disminuir la velocidad de infusión a la mitad.
- Difenhidramina 50 mg, vía oral o intravenoso.
- Si hay síntomas respiratorios utilizar Beta 2 agonistas inhalados (salbutamol)
- Si persisten los síntomas respiratorios administrar adrenalina 1:1000, 0.30-0.50 ml subcutánea en extremidad superior (contraindicado en cardiópatas).
- Hidrocortisona 100 mg intravenoso y/o antipiréticos.
- Detener la infusión si los síntomas aumentan en severidad o persisten.
C. Reacciones severas, definidas como la presencia de:
• Disnea severa.
• Obstrucción de vía aérea.
• Arritmias.
• Hipotensión.
• Colapso cardiovascular.
Conducta recomendada:
- Detener inmediatamente la infusión.
- Administrar adrenalina 1:1000, 0.30-0.50 ml subcutánea en extremidad superior.
- Difenhidramina 50 mg intravenoso.
- Hidrocortisona 100 mg intravenoso.
- Para síntomas respiratorios utilizar beta B2 agonistas inhalados o en nebulización.
- Si hay disnea severa con cianosis o sibilancias colocar oxígeno por máscara o bigotera.
- Manejar volumen de fluidos.
- En caso de paro cardio-respiratorio iniciar maniobras de reanimación cardiopulmonar.
Agradecimientos:
Los autores agradecen la colaboración de Sanofi-Genzyme para la realización de estas recomendaciones, declarando no haber recibido honorarios ni haber sido influenciados al momento de la preparación del manuscrito.
 Tabla 1
Tabla 1